Disease Testing
Tandem Mass Spectrometry can be used for disease testing. Detectable disorders include metabolic disorders, hematologic disorders, and endocrinopathies. These three groups of disorders account for approximately 3,000 new cases of potentially fatal or debilitating disease each year for which outcomes are improved with early identification and treatment through newborn screening systems. See also: "A Layperson's Guide to Tandem Mass Spectrometry and Newborn Screening" (PDF)
Amino Acid Disorders
| Disease | Argininemia |
| Metabolite | Cit and Arg |
| Incidence | 1 in 300,000 to 1 in 1,000,000 |
| Symptoms | Muscle stiffness, developmental delay and mental retardation. |
| Treatment | A low protein diet to prevent arginine and ammonia build up and there are medications that help the body get rid of excess arginine and ammonia |
| Age of Onset | 2 months to 4 years old |
| Animal Equivalents | Afects the final stage in the urea cycle in the liver so therefore all mammals are potential victims of Argininemia. (Ureatelic animals, animals with the urea cycle.) |
| Metabolite Concentration Level Range | Arginine and citrulline elevated. Arg - 152-1756uM |
| Prognosis | Good prognosis if disorder is treated prospectively from birth, progressive neurological manifestations if untreated |
| Metabolite + Missing or Ineffective Enzyme → Result | Arginine + arginase enzyme → ammonium, arginine, citrulline, lysine, and ornithine levels rise 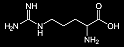 (Arginine) |
| Disease | Argininosuccinic Aciduria (ASA) |
| Metabolite | Cit |
| Incidence | 1 in 70,000 |
| Symptoms | Lack of appetite, vomiting, listlessness, seizures, and coma |
| Treatment | Treatment may include a high-caloric, protein-restrictive diet, arginine supplementation, administration of sodium benzoate and sodium phenylacetate. It may be necessary to prescribe dialysis in some cases. |
| Age of Onset | Disease is present at birth but symptoms aren't present until days or weeks after birth |
| Animal Equivalents | Potentially all Mammals |
| Metabolite Concentration Level Range | Patient has condition if Cit <51 µmol/L |
| Prognosis | Natural History without Treatment: Mental and physical retardation due to hyperammonemia, cylic vomiting, seizures, cerebal edema and trichorrhexis nodosa. Coma and death possible. Natural History with Treatment: Normal mental and physical development is possible if treatment is initiated before hyperammonemic crisis. |
| Metabolite + Missing or Ineffective Enzyme → Result | Argininosuccinate+ Argininosuccinase → ammonia, citrulline, glutamine, and urine argininosuccinic acid buildup 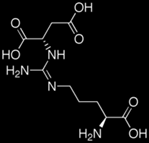 (Arginine) |
| Disease | Carbamoyl Phosphate Synthetase I Deficiency (CPS1) |
| Metabolite | Nitric Oxide (NO) |
| Incidence | 1 in 800,000 |
| Symptoms | Lethargic, unwilling to eat, loss of breathing rate control, high body temperature, seizures (and possible coma), and possible developmental delay and intellectual disability. |
| Treatment | Valine, leucine, isoleucine, methionine and phenylalanine supplements |
| Age of Onset | Days from Birth |
| Animal Equivalents | Human, Rat, and Rabbit |
| Metabolite Concentration Level Range | Patient has condition if Nitric Oxide 14.0 µmol/L higher than normal 8.5 µmol/L lower than normal |
| Prognosis | 100% mortality if untreated, there is a direct correlation between the duration of hyper-ammonemic coma and morbidity (mental retardation, developmental delays, cortical atrophy), and good prognosis if disorder is treated prospectively from birth |
| Metabolite + Missing or Ineffective Enzyme → Result | NH=O+ |
| Disease | Citrullinemia (Type I and Type II) |
| Metabolite | Cit |
| Incidence | 1 in 100,000 |
| Symptoms | Increased blood ammonia level (type I), Poor appetite (type I), Listlessness (type I), Vomiting (type I), Seizures (type II), Lack of argininosuccinate in blood (type I), Increased blood levels of citrulline (type I), Increased levels of ammonia in body tissues (type I), Increased citrulline levels (type I and II), Confusion (type II), Memory loss (type II), Restlessness (type II), Abnormal behaviors (type II), Irritability (type II), Hyperactivity (type II), Aggression (type II), Impaired vision (late onset form), Severe headache (late onset form), Balance problems (late onset form), Incoordination (late onset form), Reduced energy (late onset form) |
| Treatment | Having a low-protein diet and/or special medical foods and formula is necessary to keeping the ammonia levels down. There are medications that are taken by mouth or by tube feeding to prevent high ammonia levels. There are also medications to help remove existing ammonia in the body. Dialysis is an example of a medication that helps remove ammonia from the blood. Also, an amino acid called arginine is often given by mouth to help prevent ammonia build-up. |
| Age of Onset | Less than 30 days old |
| Metabolite Concentration Level Range | Patient has condition if Cit >51 µmol/L |
| Prognosis | Timely diagnosis and treatment usually results in a good prognosis with normal growth and learning abilities - treatment is lifelong. In some severe cases, high ammonia levels can cause complications even with treatment. |
| Metabolite + Missing or Ineffective Enzyme → Result | Citrulline + argininosuccinic acid synthetase › Citrulline and ammonia levels increase |
| Disease | Homocystinuria (Cystathione Synthase Deficiency) |
| Metabolite | Met |
| Incidence | 1 in 82,000 |
| Symptoms | Symptoms include mental retardation, seizures, psychiatric disturbances, delays in reaching developmental milestones (e.g., crawling, walking, sitting), displacement of the lens of the eye (ectopia lentis), abnormal thinning and weakness of the bones (osteoporosis and scoliosis ), and/or the formation of blood clots (thrombi) in various veins and arteries that may lead to life-threatening complications. |
| Treatment | Homocystinuria is treated initially by changing the baby to a formula that does not contain methionine. Treatment may also include a methionine-restricted and cystine-supplemented diet, as well as large doses of Vitamin B6. |
| Metabolite Concentration Level Range | Patient has condition if Met <96 |
| Age of Onset | 3-4 years old |
| Prognosis | Although no cure exists for homocystinuria, vitamin B6 therapy can help about half of people affected by the condition. If the diagnosis is made while a patient is young, starting a low methionine diet quickly can prevent some mental retardation and other complications of the disease. For this reason, some states screen for homocystinuria in all newborns.Patients with persistent rises in blood homocysteine levels are at increased risk for blood clots. Clots can cause significant medical problems and shorten lifespan. |
| Disease | Hypermethioninemia |
| Metabolite | Met |
| Incidence | 1 in 60,000 |
| Symptoms | There are usually no visible symptoms |
| Treatment | Prescribe intense methionine supplements; increase the levels of Vitamin B6 in diet, while decreasing the levels of protein in one's diet. |
| Age of Onset | The metabolic disorder is present at birth but symptoms usually arrive later in childhood. |
| Metabolite Concentration Level Range | Patient has condition if Condition when Met >960 |
| Prognosis | Excellent with initiative of treatment shortly after birth |
| Disease | Hyperammonemia (Hyperornithinemia, Homocitrullinuria Syndrome) |
| Metabolite | Orn |
| Incidence | Unknown (Only 50 Cases Reported Internationally) |
| Symptoms | Developmental delay, seizures, retarded growth, learning disability , and even neonatal death |
| Treatment | Ornithine and arginine supplementation in patient's daily diet proved to lower ammonia levels in the body |
| Age of Onset | At Birth |
| Metabolite Concentration Level Range | Patient has condition if Orn <345 |
| Prognosis | 100% mortality if untreated, there is a direct correlation between the duration of hyper-ammonemic coma and morbidity (mental retardation, developmental delays, cortical atrophy), and good prognosis if disorder is treated prospectively from birth |
| Disease | Ketotic Hyperglycinemia |
| Metabolite | Gly |
| Incidence | Unknown |
| Symptoms | Ketoacidosis, low blood sugar, intense hunger, headaches, loss or weakening of the body's basic motor functions, convulsions and loss of focus, as well as ketosis (a state in metabolism wherein the liver converts fat into fatty acids and ketone bodies that are normally used by the body as an alternative energy source. Also the level of ketone itself may slowly decarboxylate into acetone, which may be toxic to the body. |
| Treatment | Extended fasts should be avoided. The child should be given a bedtime snack of carbohydrates and should be awakened and fed after the usual duration of sleep. If the child is underweight, a daily nutritional supplement may be recommended. If a spell begins, carbohydrates and fluids should be given promptly. If vomiting prevents this, the child should be taken to the local emergency department for a few hours of intravenous saline and dextrose. |
| Age of Onset | Within hours of birth |
| Metabolite Concentration Level Range | 60 mg per deciliter or less is highly suspicious of hypoglycemia |
| Prognosis | 100% mortality if untreated but if treatment initiation is shortly after birth, patient exhibits minimum symptoms with probable chance of developing normal |
| Disease | Nonketotic Hyperglycinemia (NKH) |
| Metabolite | Gly |
| Incidence | 1 in 55,000 |
| Symptoms | Neurological symptoms are muscular hypotonia, seizures, respiratory distress, and lethargy due to elevated levels of glycine in the cerebrospinal fluid (CSF) |
| Treatment | Treatment is difficult because the removal of glycine levels in the cerebrospinal fluid by itself is not to avoid symptoms. However, doses of diazepam and dextromethorphan have proved to be beneficial through minimizing the intensity and frequency of the symptoms |
| Age of Onset | Within hours of birth |
| Metabolite Concentration Level Range | Patient has condition if Gly < 1200 µmol/L. The definition of hypoglycemia depends on the age of the child. <55 mg/dl in children <35-45 mg/dl in neonates. The diagnosis of nonketotic hyperglycinemia is considered to depend upon the presence of increased cerebrospinal fluid glycine and an increased cerebrospinal fluid to plasma glycine ratio. |
| Prognosis | Unknown |
| Disease | Hyperornithinemia with Gyral Atrophy (HOGA) |
| Metabolite | Orn |
| Incidence | Unknown |
| Symptoms | Decreased night vision, complete blindness around 50 years of age, mild muscle weakness, but patients is usually overall develop normally |
| Treatment | A few gyrate atrophy patients will respond to pharmacologic doses of vitamin B6 (pyridoxine) with increase in residual enzyme activity, partial reduction in plasma Ornithine and stabilization of vision. The slow progression of the degenerative changes in vision and the difficulty in measuring small changes in ocular function make evaluation of any therapy difficult. Additional approaches to therapy may be efficacious, including dietary reduction in Ornithine and administration of creatine |
| Age of Onset | Within days of birth |
| Metabolite Concentration Level Range | Patient has condition if Orn <345 µmol/L |
| Prognosis | Unknown |
| Disease | Maple Syrup Urine Disease (MSUD) |
| Metabolite | Leu, Leu/Ala, Val |
| Incidence | 1 in 185,000 |
| Symptoms | Loss of appetite, vomiting, poor weight gain, increasing lethargy (difficult to wake up), characteristic burned sugar smell to urine, changes in muscle tone, muscle spasms, and seizures |
| Treatment | The main treatment for maple syrup urine disease is restriction of dietary forms of the three amino acids leucine, isoleucine, and valine. These restrictions must be lifelong. There are several commercial formulas and foods for individuals with MSUD. |
| Age of Onset | Infants 4-7 days old |
| Metabolite Concentration Level Range | Patient has condition if Val <273 µmol/L, Leu <320 µmol/L |
| Prognosis | This disease can be life threatening if untreated. Even with dietary treatment, stressful situations and illness can still cause high levels of certain amino acids. Death may occur during these episodes. With strict dietary treatment, children have grown into healthy adulthood. |
| Disease | Ornithine Transcarbamylase Deficiency (OTC) |
| Metabolite | Orn |
| Incidence | 1 in 80,000 |
| Symptoms | Anorexia, Irritability, Heavy or rapid breathing, Lethargy, Vomiting, Disorientation, Somnolence, Asterixis (rare), Combativeness, Obtundation, Coma, Cerebral edema or even Death (if treatment is not forthcoming or effective) |
| Treatment | High Carbohydrate and Protein restricted diet, arginine supplements, and Sodium phenylacetate and sodium benzoate |
| Age of Onset | Earliest as disorder present at birth |
| Prognosis | Timely diagnosis and treatment usually results in a good prognosis with normal growth and learning abilities - treatment is lifelong. In some severe cases, high ammonia levels can cause complications even with treatment. |
| Disease | 5-Oxoprolinuria (Pyroglutamic Aciduria) |
| Metabolite | 5-Oxopro |
| Incidence | Unknown |
| Symptoms | Metabolic acidosis, hemolytic anemia, jaundice and urinary excretion of large amounts of 5-oxoproline in body. Neurological symptoms such as ataxia, spasticity and/or seizures may result from chronic metabolic acidosis, if not treated. |
| Treatment | Early diagnosis and prompt treatment is essential for an improved prognosis. Individuals with GSD require prompt correction of any metabolic acidosis and/or hyperbilirubinemia to help prevent brain damage. Oral maintenance doses of sodium bicarbonate or citrate may help to correct chronic acidosis. Anemia may require transfusions and any electrolyte imbalances should be corrected. Patients with generalized GSD may have heightened sensitivity to oxidative stress, and large doses of vitamin E and vitamin C may be indicated Aggressive medical management is necessary during any intercurrent illness. |
| Age of Onset | At Birth |
| Prognosis | Unknown |
| Disease | Phenylketonuria (PKU) |
| Metabolite | Phe, Phe/Tyr |
| Incidence | 1 per 13,500 to 1 per 19,000 |
| Symptoms | Left untreated, this condition can cause problems with brain development, leading to progressive mental retardation, brain damage, and seizures. |
| Treatment | However, PKU is one of the few genetic diseases that can be controlled by diet. A diet low in phenylalanine and high in tyrosine can be a very effective treatment. There is no cure. Damage done is irreversible so early detection is crucial. |
| Age of Onset | Less than 1 year old |
| Metabolite Concentration Level Range | Patient has condition if Phe <132 µmol/L |
| Prognosis | Excellent with initiation of treatment shortly after birth |
| Disease | Tyrosinemia (Type II and Type III) |
| Metabolite | Tyr/Met |
| Incidence | 1 in 100,000 |
| Symptoms | Acute Form - poor appetite and failure to grow normally, vomiting, diarrhea, bloody stools, a cabbage-like odor, jaundice (yellow skin and whites of eyes), swollen liver, irritability, and lethargy (overwhelming tiredness). Chronic Form - Cirrhosis of the liver; pain, numbness, and tingling in parts of the body (polyneuropathy), kidney problems, and episodes of intense abdominal pain. |
| Treatment | Restrict the amount of tyrosine and phenylalanine in the baby's food. Nitisinone reduces the toxic effects of tyrosine in the body. The medication, when used along with the dietary restrictions, has been successful in reducing the symptoms of tyrosinemia type I. The last method of treatment is a liver transplant. Now, thanks to nitisinone, surgery is reserved for severe cases of liver damage or cancer. However transplantation carries many risks with it, including the rejection of the new liver by the body. |
| Age of Onset | Less than 1 year of age |
| Metabolite Concentration Level Range | Patient has condition if Tyr >336 µM tyrosine (or >9.1 mg/dL) |
| Prognosis | When treatment is started early, severe liver, kidney, and neurologic symptoms can be prevented. Children who are treated usually have normal growth and intelligence. If treatment is not started right away, children may have some liver or kidney damage. Rickets may already be present and need to be treated. Delays in growth and development may also be present. The effects of delayed treatment vary from child to child. |
Fatty Acid Oxidation Disorders
| Disease | 2, 4-Dienoyl-CoA Reductase Deficiency (DE RED) |
| Metabolite | C10:2 |
| Incidence | 1 in 100,000 |
| Symptoms | Dysmorphism, early death, feeding difficulties, poor feeding, hypotonia, microcephaly, respiratory acidosis and vomiting |
| Treatment | A formula containing fat derived from medium-chain triglycerides (MCT), administering pharmacologic doses of carnitine, and avoiding fasting |
| Age of Onset | Disorder present at birth |
| Animal Equivalents | Unknown |
| Metabolite Concentration Level Range | Patient has condition if C10:2 <.1 µmol/L |
| Prognosis | Unknown |
| Metabolite + Missing or Ineffective Enzyme → Result | Unknown |
| Disease | Neonatal Carnitine Palmitoyltransferase Type I Deficiency |
| Metabolite | C0 |
| Incidence | 1 in 100,000 |
| Symptoms | Hypoketotic hypoglycemia, Encephalopathy, Enlarged liver, High blood level of carnitine, Mild metabolic acidosis, Lactic academia, Hyperammonemia, Increased transaminases, Low blood sugar, Loss of consciousness, Seizures, Muscle weakness, Nervous system damage. |
| Treatment | Avoid fasting, MCT oil supplement and avoid dietary long-chain fatty acids. |
| Age of Onset | 8-18 months from birth |
| Metabolite Concentration Level Range | Patient has condition if C0 >100 |
| Natural History Without Treatment | While most patients have survived infancy and acute hypoglycemic episodes, some have suffered permanent neurological deficits. Death has occurred during acute episodes in some cases. |
| Natural History With Treatment | Theoretically normal development if hypoglycemia and cardiac involvement can be prevented. |
| Disease | Carnitine Uptake Defect (CUD) (Carnitine Transport Defect) |
| Metabolite | C0 |
| Incidence | 1 in 250,000 |
| Symptoms | Poor appetite, vomiting, irritability, lethargy, hypoketotic hypoglycemia, anemia, metabolic acidosis, hypoglycemia, muscle weakness, cardiomyopathy, hepatomegaly, seizures brain injury from hypoglycemia. |
| Treatment | Carnitine supplementation, avoid fasting, and possible glucose feeding through an IV when patient has a viral illness |
| Age of Onset | Disorder present at birth |
| Metabolite Concentration Level Range | Patient has condition if C0 <6.0 µmol/L |
| Prognosis | The prognosis is poor during infancy but prognosis is good if the patient survives the neonatal period. |
| Metabolite + Missing or Ineffective Enzyme → Result | Carnitine + Carnitine transporter enzyme—> Urinary carnitine level increases |
| Disease | Short-chain Acyl-CoA Dehydrogenase Deficiency (SCAD) |
| Metabolite | C4 |
| Incidence | 1 in 40,000 to 1 in 100,000 |
| Symptoms | Infants will exhibit vomiting, low blood sugar (hypoglycemia), a lack of energy (lethargy), poor feeding, and failure to gain weight and grow at the expected rate (failure to thrive). Other features of this disorder may include poor muscle tone (hypotonia), seizures, developmental delays, and a small head size (microcephaly). |
| Treatment | Avoiding triggers such as periods of fasting and viral infections, feeding glucose through an IV, and adding Carnitine to infants daily diet. |
| Age of Onset | Infancy |
| Metabolite Concentration Level Range |
Patient has condition if C4 <1.24 C4 (butyryl/ isobutyryl carnitine) - elevated. C4/C2 - elevated C4/C3 - elevated |
| Prognosis | There is no proven efficacy of the usual treatments for fatty acid oxidation disorders, with the possible exception of avoiding fasting. |
| Disease | Medium-chain Acyl-CoA Dehydrogenase Deficiency (MCAD) |
| Metabolite | C8, C10, C10:1, C6 |
| Incidence | 1 in 12,000 (average) |
| Symptoms (Disease Description) | Medium-chain acyl-coenzyme A dehydrogenase is one of the enzymes involved in mitochondrial fatty acid Β-oxidation, which fuels hepatic ketogenesis, a major source of energy once hepatic glycogen stores become depleted during prolonged fasting and periods of higher energy demands. In a typical clinical scenario, a previously healthy child with MCAD deficiency presents with hypoketotic hypoglycemia, vomiting, and lethargy triggered by a common illness. Seizures may occur. Such an episode may quickly progress to coma and death. Hepatomegaly and acute liver disease are often present. |
| Treatment | Preventing from fasting longer than 12 hours, having a low-fat diet (e.g., <30% of total energy from fat), administration of glucose to prevent hypoglycemia. |
| Age of Onset | 3 months to 24 months |
| Metabolite Concentration Level Range | Patient has condition if C6 <.26 µmol/L, C8 <.46 µmol/L, C10:1 <.35 µmol/L |
| Prognosis | Excellent when treated before the onset of symptoms |
| Disease | Very-long-chain Acyl-CoA Dehydrogenase Deficiency (VLCAD) |
| Metabolite | C14:1, C14, C16, C18 |
| Incidence | 1 in 40,000 to 1 in 120,000 |
| Symptoms | Low blood sugar (hypoglycemia), lack of energy (lethargy), and muscle weakness, liver abnormalities and life-threatening heart problems. Symptoms that begin in adolescence or adulthood tend to be milder and usually do not involve heart problems. Episodes of very long-chain acyl-coenzyme A dehydrogenase deficiency can be triggered by periods of fasting, illness, and exercise. |
| Treatment | Preventative methods include feeding patients glucose through an IV as an energy source, monitoring for cardiac rhythm disturbance, and monitoring for rhabdomyolysis, avoidance of triggers, such as fasting, long chain fats, and irritation of the myocardium).Cardiac dysfunction is reversible with early, intensive supportive care (occasionally including extra-corporeal membrane oxygenation) and diet modification. |
| Age of Onset | Infancy |
| Metabolite Concentration Level Range | Patient has condition if C14 <.62 µmol/L, C14:1 <.54 µmol/L, C16 <8.35 µmol/L, C18 <2 µmol/L |
| Prognosis | Most infants with condition die within the first few months, however long-term survival has been reported in infants with a strictly controlled diet |
| Disease | Multiple Acyl-CoA Dehydrogenase Deficiency (MADD) |
| Metabolite | C4, C5, C8:1, C8, C12, C14:1, C16, C5-DC, C10 |
| Incidence | Unknown |
| Symptoms | Newborns may have abnormal features, such as wide spaced eyes and low set ears, genital abnormalities, rocker-bottom feet. They may also have low muscle tone, low blood sugar, large livers, and have the smell of sweaty feet. However sometimes people with GA-II may not have symptoms until later in life. These symptoms may be low blood sugar, liver disease, and neurological problems. |
| Treatment | There is no effective treatment for the severe forms of MADD that present in the neonatal period. Patients with later onset less severe symptoms may respond to riboflavin (a precursor to FAD) and carnitine supplementation. Dietary restriction of fats and protein has had variable results. |
| Age of Onset | Disorder present at birth, however symptoms possibly may not arrive until adulthood |
| Metabolite Concentration Level Range | Patient has condition if C4 <1.24, C5 <.81 µmol/L, C5-DC < .29 µmol/L, C10 <.41 µmol/L, C14:1 <.54 µmol/L, C16 <8.35 µmol/L |
| Prognosis | Most infants with structural abnormalities die within the first week of life. The few that respond to initial treatment often succumb to cardiomyopathy in infancy. |
| Disease | Carnitine Palmitoyl Transferase Deficiency Type I (Liver) (CPTI) |
| Metabolite | C16, C18:1 |
| Incidence | Unknown (Only 50 cases identified) |
| Symptoms | Low levels of ketones (which are products of fat breakdown that are used for energy), low blood sugar (hypoglycemia) is another major feature. These two conditions lead the patient to be diagnosed with hypoketotic hypoglycemia, which can result in a loss of consciousness or seizures. People with this disorder typically also have an enlarged liver (hepatomegaly), muscle weakness, nervous system damage, and elevated levels of carnitine in the blood. Carnitine is usually a natural substance acquired mostly through the diet. Carnitine is required by cells to process fats and produce energy. People with this disorder have a faulty enzyme that disrupts carnitine's role in processing long-chain fatty acids. |
| Treatment | Sufficient amounts of intravenous 10% dextrose should be provided as quickly as possible to correct hypoglycemia and to prevent lipolysis and subsequent mobilization of fatty acids into the mitochondria. A high-carbohydrate diet (70% of calories) that is low in fat (<20% of calories) is generally recommended to provide a constant supply of carbohydrate energy, particularly during illness. Provision of approximately one-third of total calories as medium-chain triglycerides is recommended. C6-C10 fatty acids do not require the carnitine shuttle for entry into the mitochondrion. Frequent feeding is recommended, particularly for infants, given their limited glycogen reserves. Cornstarch feedings given overnight provide a constant source of slow-release carbohydrate to prevent hypoglycemia during sleep. Older children should not fast for more than 12 hours and for a shorter time if evidence of a febrile or gastrointestinal illness exists. |
| Age of Onset | Disorder Present at Birth |
| Metabolite Concentration Level Range | Patient has condition if C16 >.4 µmol/L |
| Prognosis | The prognosis for infants with a neonatal presentation is poor with overall mortality in the first episode as high as 60%. However patients who survive to diagnosis have a good prognosis. |
| Metabolite + Missing or Ineffective Enzyme → Result | Transport Fatty Acids + carnitine palmitoyl transferase —> elevated C16 and C18 resulting in rapid onset of hypoglycemic crises. |
| Disease | Carnitine Palmitoyl Transferase Deficiency Type II |
| Metabolite | C16 |
| Incidence | Unknown |
| Symptoms | Aching muscles, Fatigue, Breakdown of muscle tissue, myoglobins in the urine after cold exposure, myoglobins in the urine after fever. |
| Treatment | Patients with adult-onset muscle form of the disease must alter their lifestyle and refrain from rigorous exercise. It is probably prudent to avoid prolonged fasting. Medium-chain triglyceride oil may be beneficial for all patients, because it bypasses the need for CPT II activity. Aggressive treatment of acutely ill infants with IV glucose and cardiac support is critical. L-Carnitine supplementation should be instituted. Any intercurrent infection or illness will be life threatening to patients affected with the childhood form. |
| Age of Onset | Disorder present at infancy |
| Metabolite Concentration Level Range | Patient has condition if C16 <8.35 µmol/L |
| Prognosis | Condition is fatal in most patients during infancy but patients who survive to diagnosis have a good prognosis |
| Disease | Carnitine/Acylcarnitine Translocase Deficiency (CACT) |
| Metabolite | C16, C18:1, C18 |
| Incidence | Unknown (Less than 30 cases identified) |
| Symptoms | Seizures, an irregular heartbeat (arrhythmia), and breathing problems, an extremely low level of ketones in the body (which are products of fat breakdown that are used for energy), and low blood sugar (hypoglycemia). Together these signs are called hypoketotic hypoglycemia, which can result in unconsciousness and seizures. Other signs that are often present include excess ammonia in the blood (hyperammonemia), an enlarged liver (hepatomegaly), heart abnormalities (cardiomyopathy), and muscle weakness. |
| Treatment | Providing sufficient amounts of glucose is essential to prevent collapse, brain damage, cardiac arrhythmia, and sudden death. |
| Age of Onset | Hours after Birth |
| Metabolite Concentration Level Range | Patient has condition if C16 <8.35 µmol/L, C18 < 2.0 µmol/L, C18:1 <3.60 µmol/L, C18:2 <2.1 µmol/L |
| Prognosis | Prognosis is good if patient receives treatment before the onset of symptoms |
| Disease | Short-Chain Hydroxyacyl-CoA Dehydrogenase Deficiency (SCHADD) |
| Metabolite | C4, C16 |
| Incidence | Unknown |
| Symptoms | Extreme sleepiness, behavior changes, irritable mood, poor appetite, fever, diarrhea, vomiting, hypoglycemia, breathing problems, seizures, swelling of the brain, coma, sometimes leading to death, irregular heart beat and other heart problems, enlarged heart, liver problems, muscle problems, high levels of insulin in the blood in some babies. |
| Treatment | Diazoxide supplements, avoid fasting, L-carnitine supplements, and a low fat/ high carbohydrate diet. |
| Age of Onset | Disorder present at birth |
| Prognosis | Natural History without Treatment: Extremely variable. Neonatal presentation consisted of difficulty with feeding, failure to thrive, hypotonia and an elevated CPK. Others presented with recurrent myoglobinuria and hypoketotic hypoglycemia. Natural History with Treatment: Treatment should prevent liver problems. The effectiveness of treatment in managing other symptoms is not known. Treatment will not reverse CNS damage due to hypoglycemic episodes. |
| Disease | Long-Chain Hydroxyl Acyl-CoA Dehydrogenase Deficiency (LCHAD) |
| Metabolite | C16-OH, C18, C18:1-OH, C14-OH, C16 |
| Incidence | Unknown |
| Symptoms | Muscle weakness, low blood sugar, and lack of energy |
| Treatment | Prevention of hypoglycemia with frequent feedings seems appropriate. Fasting should be avoided, particularly during times of illness. Dietary supplementation with uncooked food-grade cornstarch after the first year or two of life should be considered, because it may permit longer periods of normoglycemia. Those patients with documented hyperinsulinism have responded to treatment with diazoxide. High carbohydrate intake should be encouraged during illness, with initiation of intravenous glucose supplementation if the child is unsuccessful in keeping down fluids, or unable to take adequate oral feedings. |
| Age of Onset | Disorder present at birth |
| Metabolite Concentration Level Range | Patient has condition if C14-OH< .11 µmol/L, C16 <8.35 µmol/L, C16-OH < .13 µmol/L, C18 < 2 µmol/L, C18:1-OH <.08 µmol/L |
| Prognosis | Prognosis, as well as treatment, is poor for this disorder. However patients who respond to riboflavin supplementation are expected to live longer |
| Disease | Trifunctional Protein Deficiency (TFP Deficiency) |
| Metabolite | C16-OH, C18, C18OH, C16 |
| Incidence | 1 in 100,000 |
| Symptoms | Feeding difficulty, lethargy, lack of energy, low blood sugar, muscle weakness, and liver problems |
| Treatment | An avoidance of fasting and aggressive medical management during illness, and not receiving adequate nutritional intake are all essential to the survival of the patient. At the time of illness, the infant/child should be admitted for medical care, including administration of intravenous dextrose to prevent hypoglycemia. Supplemental carnitine, a low-fat diet and home glucose monitoring may also be necessary. |
| Age of Onset | Disorder present at birth |
| Metabolite Concentration Level Range | Patient has condition if C16 <8.35 µmol/L, C16-OH < .13 <mol/L, C18< 2 µmol/L |
| Prognosis | Early TFP deficiency: Most babies with early TFP deficiency die of heart or breathing problems, even when treated. However, treatment may help prolong life in some babies. Childhood TFP deficiency: With prompt and careful treatment, children with TFP deficiency can often live healthy lives with typical growth and development. However, some children continue to have episodes of hypoglycemia or metabolic crisis, even with treatment. This can cause permanent brain damage and may result in learning disabilities or mental retardation. Mild/muscle TFP deficiency: When treated, people with mild/muscle TFP deficiency usually remain healthy. This form does not affect intelligence. |
| Disease | Mitochondrial Acetoacetyl CoA Thiolase Deficiency (ACAT) or Beta Ketothiolase (BKT) |
| Metabolite | C5:1, C5-OH |
| Incidence | Unknown |
| Symptoms | A special low-protein diet that limits isoleucine intake and avoids fasting. Some doctors may also prescribe dietary supplements. Emergency care must be taken if a person with BKT deficiency becomes ill and has difficulty keeping food down. This is usually treated in the hospital with an IV. |
| Treatment | Plasma levels of glucose, electrolytes, and ammonia should be normalized. Carnitine supplementation may be helpful. For the long-term, affected patients should avoid fasting, eat frequently, and restrict protein intake. Intravenous glucose can be used when the patient is febrile or vomiting. Carnitine supplementation is reasonable. With appropriate monitoring and therapy, there is a good prognosis for normal development. |
| Age of Onset | 3 days from birth to 48 months old |
| Metabolite Concentration Level Range | Patient has condition if C5:1 <.07 µmol/L, C5-OH <.86 µmol/L |
| Prognosis | There is no cure for beta ketothiolase deficiency. However, the outcome is usually good in infants who are treated early and continue with lifelong treatment. |
| Disease | Medium-chain Ketoacyl-CoA Thiolase Deficiency (MCKAT) |
| Metabolite | C12-C16 |
| Incidence | Unknown |
| Symptoms | vomiting, dehydration, metabolic acidosis, liver dysfunction, and terminal rhabdomyolysis with myoglobinuria |
| Treatment | Unknown |
| Age of Onset | Before 5 months old |
| Prognosis | Unknown (Fatal if untreated) |
Organic Disorders
| Disease | Ethylmalonic Encephalopathy (EME) |
| Metabolite | C4, C5, and C2 |
| Incidence | Unknown (Less Than 30 Cases Reported) |
| Symptoms | Progressively delayed development, weak muscle tone (hypotonia), seizures, and abnormal movements. The body's network of blood vessels (the vascular system) is also affected. Children with this disorder may experience rashes of tiny red spots (petechiae) caused by bleeding under the skin and blue discoloration in the hands and feet due to reduced oxygen in the blood (acrocyanosis). Chronic diarrhea is another common feature of ethylmalonic encephalopathy. |
| Treatment | L-carnitine and riboflavin supplements may be recommended |
| Age of Onset | Disorder present at birth |
| Metabolite Concentration Level Range | Carnitine range 21-53 µmol/L with elevated C4, C5, and C2 levels |
| Prognosis | The signs and symptoms of ethylmalonic encephalopathy are apparent at birth or begin in the first few months of life. Problems with the nervous system typically worsen over time, and most affected individuals survive only into early childhood. |
| Disease | Glutaric Acidemia Type I (GA-1) (Glutaryl-CoA Dehydrogenase Deficiency) |
| Metabolite | C5-DC |
| Incidence | 1 in 30,000 to 1 in 40,000 |
| Symptoms | This is a disease where inadequate levels of an enzyme that helps break down the amino acids lysine, hydroxylysine, and tryptophan. Intellectual disability may also occur. The severity of glutaric acidemia type I varies widely; some individuals are only mildly affected, while others have severe problems. In most cases, signs and symptoms first occur in infancy or early childhood, but in a small number of affected individuals, the disorder first becomes apparent in adolescence or adulthood. Some babies with glutaric acidemia type I are born with unusually large heads (macrocephaly). Affected individuals may have difficulty moving and may experience spasms, jerking, rigidity, or decreased muscle tone. Some individuals with glutaric acidemia have developed bleeding in the brain or eyes that could be mistaken for the effects of child abuse. |
| Treatment | Frequent feeds (4-6hrly); prevention of fasting, including overnight. Uncooked cornstarch, which releases its glucose very slowly through the night, may help. In severe cases some restrict fat intake. |
| Age of Onset | Disorder present at birth |
| Metabolite Concentration Level Range | Patient has condition if C5-DC <.29 µmol/L |
| Prognosis | Possible developmental delay due to encephalitis-like crisis; neurologic deterioration including spasticity, dystonic cerebral palsy. May have neurologic signs with normal IQ. Some individuals may be asymptomatic. |
| Metabolite + Missing or Ineffective Enzyme → Result | Lysine + glutaryl-CoA dehydrogenase → C5-DC, Glutaric, and 3-OH-glutaric acid level increases |
| Disease | Isobutyryl CoA Dehydrogenase Deficiency (IBCD) |
| Metabolite | C4 |
| Incidence | Unknown (Less than 5 cases have been reported) |
| Symptoms | Poor feeding and growth (failure to thrive), a weakened and enlarged heart (dilated cardiomyopathy), seizures, and low numbers of red blood cells (anemia), very low blood levels of carnitine (a natural substance that helps convert certain foods into energy). Symptoms may be worsened by long periods without food (fasting) or infections that increase the body's demand for energy. Some individuals with gene mutations that can cause isobutyryl-CoA dehydrogenase deficiency may never experience any signs and symptoms of the disorder. |
| Treatment | Carnitine therapy reversed the cardiomyopathy. Moderate protein restriction to reduce valine intake and avoidance of fasting is prudent. |
| Age of Onset | Disorder present at birth but symptoms arrive later in childhood |
| Metabolite Concentration Level Range | Patient has condition if C4 <1.24 µmol/L |
| Prognosis | Unknown |
| Disease | Isovaleric Acidemia (IVA) (Isovaleryl-CoA Dehydrogenase Deficiency) |
| Metabolite | C5 |
| Incidence | 1 in 100,000 to 1 in 200,000 |
| Symptoms | Poor feeding, vomiting, seizures, and lack of energy (lethargy), and a buildup of a compound called isovaleric acid |
| Age of Onset | Days from birth |
| Metabolite Concentration Level Range | Patient has condition if C5 <.81 µmol/L |
| Prognosis | Natural History without Treatment: About 50% of patients with the acute neonatal form will die during their first episode. Survivors may have neurological damage though several have made complete recoveries. Patients with the chronic form may have neurologic damage, but the majority of patients are developmentally normal. Natural History with Treatment: Intellectual prognosis depends on early diagnosis and treatment and subsequently on long-term compliance. If treated appropriately, most will have normal development. |
| Disease | Malonic Aciduria (MA) |
| Metabolite | C3-DC |
| Incidence | Unknown (Less than 20 cases reported) |
| Symptoms | All patients have had developmental delay and 20-40% have other symptoms including: Hyptonia, hypoglycemia, metabolic acidosis, cardiomyopathy, diarrhea, vomiting, ketosis, seizures, lactic acidemia, microcephaly and low cholesterol. |
| Treatment | There is limited experience in managing this rare disorder. Dietary modification to increase the amount of calories from carbohydrate relative to fat has been effective in improving metabolic abnormalities. Extended fasting should be avoided. Carnitine supplementation has been beneficial in some patients. |
| Age of Onset | 3 days from birth to 13 years old |
| Metabolite Concentration Level Range | Patient has condition if C3-DC <.19 µmol/L |
| Prognosis | Unknown |
| Disease | Methylmalonic Acidemia (MUT) |
| Metabolite | C4-DC |
| Incidence | 1 in 25,000 to 1 in 48,000 |
| Symptoms | Brain disease that gets worse (progressive encephalopathy), dehydration, developmental delays, failure to thrive, lethargy, repeated yeast infections, seizures, and vomiting |
| Treatment | Dietary restriction of total protein (Get enough to get necessary nutrients but too much leads to mental instability), dietary restriction on the four offending amino acids (valine, isoleucine, methionine and threonine), and odd-chained fats is the treatment. Cobalamin supplements may be beneficial |
| Age of Onset | Disorder present within hours of birth |
| Metabolite Concentration Level Range | Patient has condition if C4-DC <1.84 µmol/L |
| Prognosis | Natural History without Treatment: Variable depending on the enzyme defect. Some will die in the newborn period, others will survive with deficits and others will be asymptomatic. Natural History with Treatment: CblA: Good prognosis with injections of hydroxy-cobalamin (OH-cbl) which reverses biochemical and clinical abnormalities in about 90% of patients. CblB: Equal fractions of affected patients are alive and well, alive and impaired, or deceased. The age of onset of symptoms can help prognosticate outcome. Those patients with a later onset of symptoms have a more benign course. Approximately 40% of patients will respond with a drop in MMA level when given OH-cbl injections. |
| Disease | Multiple Carboxylase Deficiency (MCD) (Holocarboxyalase Synthetase or Biotinidase Deficiency) |
| Metabolite | C3, C5OH |
| Incidence | 1 in 87,000 |
| Symptoms | Seizures, hypotonia, immune system impairment, skin rashes, hair loss, hearing loss and mental retardation |
| Treatment | Oral biotin supplementation has proved to have positive effects on patients |
| Age of Onset | Disorder present at birth |
| Metabolite Concentration Level Range | Patient has condition if C3 < 7.54 µmol/L |
| Prognosis | Babies who receive prompt and ongoing treatment with biotin before they have a metabolic crisis are expected to have normal growth and development. Even with treatment, a few children have developed life-long learning problems or mental retardation. In children who have already shown delays in learning, or loss of hearing or eyesight, treatment can prevent additional effects. But, it may not be able to correct the effects that are already present. |
| Metabolite + Missing or Ineffective Enzyme → Result | Genetic inability to unbind biotin (vitamin H) from proteins |
| Disease | Propionic Acidemia (PROP) (Propionyl CoA Carboxylase Deficiency) |
| Metabolite | C3 |
| Incidence | 1 in 100,000 in U.S. |
| Symptoms | Refusal to eat, poor sucking ability, vomiting, dehydration, lethargy (excessively tired or sluggish), acidosis (excess acid in the blood), and hyperammonemia (excess ammonia in the blood) If not treated, mental impairment, coma and death can follow. |
| Treatment | Dietary restriction of total protein (Get enough to get necessary nutrients but too much leads to mental instability), dietary restriction on the four offending amino acids (valine, isoleucine, methionine and threonine), and odd-chained fats is the treatment. |
| Age of Onset | Hours from birth |
| Metabolite Concentration Level Range | Patient has condition if C3 <7.54 µmol/L |
| Prognosis | Natural History without Treatment: Metabolic crises may lead to neurologic damage including mental retardation, movement disorders, seizures. coma and sudden death are also possible. Natural History with Treatment: If treatment instituted before metabolic crisis, normal IQ and development may be seen. Treatment may improve some symptoms of affected individuals. |
| Disease | 2-Methylacetoacetic Aciduria (3-Ketothiolase or Beta Ketothiolase) |
| Metabolite | C5, C5OH |
| Incidence | 1 in 115,000- 1 in 169,000 |
| Symptoms | Episodes of metabolic acidosis and ketosis, vomiting, diarrhea, coma, possible damage at the striatal necrosis of the basal ganglia, dystonia and/or mental retardation, coma and possible death. |
| Treatment | Supply the patient with glucose and bicarconate through an IV (Usually long term), avoid rich protein and ketogenic diets, carnitine supplements may be prescribed, and to be aware of ketone levels in the body. |
| Age of Onset | 3 days to 48 months from birth |
| Metabolite Concentration Level Range | Patient has condition if C5 level is >3.7 µmol/L |
| Prognosis | Natural History without Treatment: Clinical outcome varies widely with a few patients suffering severe psychomotor retardation or death as a result of their initial attack and others with normal development and no episodes of acidosis. Natural History with Treatment: Despite severe recurrent attacks, appropriate supportive care can result in normal development. |
| Metabolite + Missing or Ineffective Enzyme → Result | Isoleucine + mitochondrial acetoacetyl-CoA thiolase → elevated C5-OH (acylcarnitine), elevated C5:1 (tiglycarnitine) |
| Disease | 2-Methylbutyrl CoA Dehydrogenase Deficiency (2MBCD) or 2-Methylbutryrl Glycinuria |
| Metabolite | C5 |
| Incidence | Unknown |
| Symptoms | Poor feeding, lack of energy (lethargy), vomiting, an irritable mood, difficulty breathing, seizures, coma, poor growth, vision problems, learning disabilities, muscle weakness, and delays in motor skills such as standing and walking. Symptoms may be triggered by prolonged periods without food (fasting), infections, or eating an increased amount of protein-rich foods. However some people with this disorder never have any signs or symptoms (asymptomatic). |
| Treatment | An amino acid dominant/protein deficient formula, use of thiamine to treat thiamine-responsive MSUD and hydroxocobalamin, but usually not cyanocobalamin to treat methylmalonic acidemia, vomiting, diarrhea, and febrile illness. |
| Age of Onset | Infancy to later in childhood |
| Metabolite Concentration Level Range | Patient has condition if C5 <.81 µmol/L |
| Prognosis | Natural History without Treament: Ranges from asymptomatic to acute neonatal decomposition with neurological deficits. The limited number of patients makes it difficult to determine the natural history of the disorder. However, the disorder is not thought to be benign in that asymptomatic individuals may not have been exposed to the environmental stressors (i.e. fasting) that can cause symptoms. Natural History with Treatment: Treatment in a symptomatic patient resolved episodic hypoglycemia but the neurologic dysfunction remains. Other patients treated from birth are asymptomatic thus far but the efficacy of the treatment remains to be established. |
| Disease | 2-Methyl-3-Hydroxybutyric Aciduria (2M3HBA) (2-methyl-3-Hydroxybutyryl CoA Dehydrogenase) |
| Metabolite | C5-OH, C5, Leu |
| Incidence | Unknown |
| Symptoms | Severe metabolic acidosis and ketosis accompanied by vomiting (often hematemesis), diarrhea and coma that may progress to death. Neurologic damage includes striatal necrosis of the basal ganglia, dystonia and/or mental retardation. Other symptoms include cardiomyopathy, prolonged QT interval, neutropenia, thrombocytopenia, poor weight gain, renal failure and short stature. |
| Treatment | Acute management of the ketoacidosis is supportive with IV glucose and bicarbonate. Bicarbonate therapy is often required long term. While protein restriction is not usually necessary, protein rich diets and ketogenic diets should be avoided. Carnitine supplementation can be used. |
| Age of Onset | 3 days to 48 months from birth |
| Metabolite Concentration Level Range | C5-OH (3-hydoxyisovaleryl carnitine) - elevated. C5:1 (methylcrotonyl or tiglyl carnitine) - elevated. Leucine- elevated. |
| Prognosis | Natural History without treatment: Uncertain, as all diagnosed patients have been treated, whether neurologicaldeterioration would progress to death. The oldest patient (also the mildest) is severely dysarthric and works in a sheltered workshop. Natural History with treatment: Uncertain what long-term therapy will show, but seemed to be some clinical improvement with dietary isoleucine restriction. |
| Metabolite + Missing or Ineffective Enzyme → Result | + Isoleucine → C5-OH,C5:1, and Leucine elevated |
| Disease | 3-Methylglutaconic Aciduria Type I |
| Metabolite | C5-OH |
| Incidence | Unknown (Less than 20 cases reported) |
| Symptoms | Speech delay, delay in the development of mental and motor skills (psychomotor delay), elevated levels of acid in the blood and tissues (metabolic acidosis), abnormal muscle tone (dystonia), and spasms and weakness of the arms and legs (spastic quadriparesis) |
| Treatment | None |
| Age of Onset | Unknown |
| Animal Equivalents | Unknown |
| Prognosis | Natural History without treatment: Varies from asymptomatic except for organic Aciduria to neurodegeneration and deafness. Natural History with Treatment: Same as for untreated group |
| Disease | 3-Methylglutaconic Aciduria Type II (3-Methylglutaconyl CoA Hydratase) A.K.A. Barth Syndrome |
| Metabolite | C5-OH |
| Incidence | Unknown (Less than 50 cases reported) |
| Symptoms | Speech delay, delayed mental development, delayed development of motor skills, metabolic acidosis, abnormal muscle tone, arm spasms, leg spasms, arm weakness, leg weakness, increased urine level of 3-methylglutaconic acid, fetal growth failure, growth failure after birth, optic atrophy, dilated cardiomyopathy, heart failure, long QT syndrome, sudden death due to heart failure, microvesicular hepatic steatosis, hypospadias, small wasted testicles, undescended testes, nonprogressive cerebellar ataxia, nonprogressive mental retardation, normochromic microcytic anemia, 3-methylglutaconic aciduria, and slightly increased liver enzymes |
| Treatment | No effective treatments |
| Age of Onset | Disorder present at birth |
| Metabolite Concentration Level Range | Patient has condition if C5-OH <.86 µmol/L |
| Prognosis | Natural History without Treatment: Without care of the neutropenia and cardiac failure patients will die. Natural History with Treatment: If care is given the patient has improved chance of survival but still at high risk for morbidity and mortality. |
| Disease | 3-Methylglutaconic Aciduria Type III (Costeff Optic Atrophy Syndrome) |
| Metabolite | C5-OH |
| Incidence | Unknown in the U.S. |
| Symptoms | Degeneration (atrophy) of the optic nerves, which carry information from the eyes to the brain. Other nervous system problems might occur, such as an inability to maintain posture, poor muscle tone, a gradual increase of involuntary jerking movements (choreiform movements), and a general decrease in brain function (cognitive deficit) |
| Treatment | None |
| Prognosis | Natural History without Treatment: Spastic paraparesis with blindness and possible mental retardation. Possibly some ataxia. Natural History with treatment: Same as for untreated group. |
| Disease | 3-Methylglutaconic Aciduria Type IV |
| Metabolite | C5-OH |
| Incidence | Unknown |
| Symptoms | Type IV patients have a disparate variety of symptoms with the only commonality being the mild excretion of 3-MGA in the urine. Patients have a clinically heterogenous and nonspecific presentation and clinical course. There is variable psychomotor retardation; hypertonicity; hypotonia; optic atrophy; dysmorphic features; seizures; cardiomyopathy and hepatic dysfunction. Others have been noted to have neurodegeneration; deafness; failure to thrive; absence of acidosis. Some patients may have Pearson syndrome- hematologic disorder, lactic acidemia, and abnormality of the electron transport chain with identified mitochondrial DNA deletions. |
| Prognosis | Natural History without Treatment: Varies from asymptomatic except for organic Aciduria to neurodegeneration and deafness. Natural History with Treatment: Same as for untreated group. |
| Disease | 3-Methylglutaconic Aciduria Type V (Dilated cardiomyopathy with ataxia) (DCMA) |
| Metabolite | C5-OH |
| Incidence | Unknown |
| Symptoms | An enlarged and weakened heart (dilated cardiomyopathy) and an inability to coordinate voluntary muscular movements (ataxia) are the hallmark signs of DCMA. Some people with DCMA can also have growth failure; mild intellectual disability; optic atrophy; and in males, undescended testes (cryptorchidism) and the opening of the urethra on the underside of the penis (hypospadias). |
| Treatment | None |
| Age of Onset | 3 years old |
| Prognosis | Natural History without Treatment: Varies from asymptomatic except for organic Aciduria to neurodegeneration and deafness. Natural History with Treatment: Same as for untreated group. |
| Disease | 3-Hydroxy-3-Methylglutaryl CoA Lyase Deficiency ((3-Hydroxy-3-Methylglutaric Aciduria (HMG-CoA lyase Deficiency)) |
| Metabolite | C5-OH |
| Incidence | Unknown |
| Symptoms | Poor appetite, extreme sleepiness or lack of energy, behavior changes, irritable mood, muscle weakness, fever, nausea, diarrhea, vomiting, hypoglycemia, increased levels of acidic substances in the blood (metabolic acidosis), high levels of ammonia in the blood, enlarged liver, breathing problems, seizures, coma, and possibly death |
| Treatment | Avoid fasting, a low-leucine diet with limited amounts of protein and fat, Some children may benefit by taking L-carnitine. During a metabolic crisis, children may be given glucose, bicarbonate, and other medications by IV to treat hypoglycemia |
| Age of Onset | Disorder present at birth but symptoms may not arrive until later in child's infancy |
| Metabolite Concentration Level Range | Patient has condition if C5-OH <.86 µmol/L, C6-OH <.20 µmol/L |
| Prognosis | With treatment, normal development and IQ are possible. However, severe episodes of hypoglycemia may still result in seizures and/or mental retardation. Without treatment, recurring metabolic crises, associated with illness or fasting, will likely result in developmental delay/mental retardation or death. Symptoms generally begin after three months and before two years of age. |
| Metabolite + Missing or Ineffective Enzyme → Result | Leucine + HMG CoA lyase → C5-OH level increases and Ketone production decreases |
| Disease | 3-Methylcrotonylglycinuria (BMCC Deficiency) (3-Methylcrotonyl CoA Carboxylase Deficiency) (3-MCC) |
| Metabolite | C5-OH |
| Incidence | 1 in 50,000 |
| Symptoms | Acute metabolic decompensation can lead to coma, lethargy, and death |
| Treatment | Leucine restricted diet with glycine and carnitine supplementation |
| Age of Onset | Around 3 months after birth |
| Metabolite Concentration Level Range | Patient has condition if C5-OH <.86 µmol/L |
| Prognosis | Natural History without Treatment: Primary manifestations appear to be muscular hypotonia and atrophy. Individuals with Reye-like illnesses may die or suffer neurologic insult during these episodes. Natural History with Treatment: Once over the initial crisis, most individuals have been intellectually normal. It is uncertain whether treatment modifies disease course. |
Other Diseases
| Disease | Congenital Hypothyroidism |
| Metabolite | Thyroid Stimulating Hormone (TSH) |
| Incidence | 1 in 3,000 to 1 in 5,000 |
| Symptoms | A large soft spot on the head (fontanel), low temperature, feeding difficulties, constipation, jaundice (yellow skin), sleepiness, or color changes. If not treated, symptoms could lead to mental retardation and permanent brain damage. Treatment: A daily prescription of thyroid hormone replacement medication |
| Age of Onset | Weeks to months after birth |
| Metabolite Concentration Level Range | Patient has condition if Thyroid Stimulating Hormone <33uU/ml <8 days of age, <13uU/ml >7 days of age |
| Prognosis | Children with CH who start treatment soon after birth usually have normal growth and intelligence and can live typical and healthy lives. Some children, even when treated, have problems with school work and may need extra help. Some may have delayed growth compared to other children their age. If treatment is not started until several months after birth, delays or learning problems may occur. The level of delay varies from child to child. |
| Metabolite + Missing or Ineffective Enzyme → Result | Thyroid stimulating hormone + Insufficient Thyroid Gland → Elevated thyroid stimulating hormone and thyroxine |
| Disease | Neuroblastoma |
| Metabolite | Catecholamine |
| Incidence | 10 in 1,000,000 |
| Symptoms | Lump in the abdomen, neck, or chest, bulging eyes, dark circles around the eyes ("black eyes"), bone pain, swollen stomach and trouble breathing in infants, painless, bluish lumps under the skin in infants, weakness or paralysis (loss of ability to move a body part) |
| Treatment | Surgery, chemotherapy, and radiation therapy |
| Age of Onset | 2 years old |
| Metabolite Concentration Level Range | Unknown |
| Prognosis | The prognosis and treatment options depend on the following: Age of the child when diagnosed. Stage of the cancer. Where the tumor is in the body. Tumor histology (the shape, function, and structure of the tumor cells). Prognosis and treatment decisions for neuroblastoma are also affected by tumor biology, which includes: The patterns of the tumor cells. How different the tumor cells are from normal cells. How fast the tumor cells are growing. The number of chromosomes in the tumor cells. How many copies of the N-myc gene there are. The tumor biology is said to be favorable or unfavorable, depending on these factors. A favorable tumor biology means there is a better chance of recovery. |
| Disease | Congenital Adrenal Hyperplasia (CAH) |
| Metabolite | 17-hydroxyprogesterone (17OHP) |
| Incidence | 1 in 15,000 to 1 in 16,000 |
| Symptoms | Abnormally large amounts of androgen (a hormone that stimulates the activity of male sex organs), abnormalities of sexual development, particularly masculinization of the external genitalia in females, deficiencies of glucocorticoids occur in some cases despite the adrenal hypertrophy, weakness, nausea, vomiting, anorexia, irritability, depression, darkening or pigmentation of the skin, hypotension, lack of resistance to cold, and inability to respond physiologically to stress, pseudohermaphroditism and symptoms could possibly lead to a fatal gluco- or mineralocorticoid deficiency crisis. |
| Treatment | Administration of enough glucocorticoid to suppress ACTH and reverse the metabolic abnormalities. A mineralocorticoid is required in the salt-losing form. Plastic surgery may be necessary in females with ambiguous genitalia. This should be performed early in life. Oral corticosteroid therapy corrects the endocrine deficiency and must continue throughout life. |
| Age of Onset | Disorder present at birth but symptoms don't arrive until patient is 3-4 years old |
| Metabolite Concentration Level Range | Patient has condition if 17-hydroxyprogesterone <1250 gmwt <135 ng/ml, 17-hydroxyprogesterone >1251 <1750 gmwt <90 ngml, 17-hydroxyprogesterone >1751 <2249 gmwt <65ng/ml, 17-hydroxyprogesterone ≥2250 gmwt <50 ng/ml |
| Prognosis | Children with CAH who start treatment soon after birth usually have normal growth and development. In most treated children, puberty occurs at the normal age, although some still have early changes. Even when treated, some adults are shorter than average. |
| Metabolite + Missing or Ineffective Enzyme → Result | 17-hydroxyprogesterone + 21 hydroxylase → 17-hydroxyprogesterone level increases |
| Disease | Galactokinase Deficiency (GALK) Galactosemia Type II |
| Metabolite | Galactose |
| Incidence | 1 in 100,000 |
| Symptoms | Infants develop cataracts, jaundice (yellowish discoloration of the skin and the whites of the eyes), vomiting, poor feeding (baby refusing to drink milk-containing formula), poor weight gain, lethargy, irritability, and convulsions |
| Treatment | Diet restriction of lactose |
| Age of Onset | Within days from birth |
| Prognosis | Though dietary therapy may decrease toxicity, CNS disease and ovarian dysfunction may still manifest. Patients may have delay in acquisition of language skills and females may have primary ovarian failure |
| Disease | Galactose Epimerase Deficiency (GALE) or Galactosemia Type III |
| Incidence | 1 in 50,000 to 1 in 65,000 |
| Symptoms | Developmental delay, motor retardation, liver disease, language retardation, learning difficulties, and concentration difficulties |
| Treatment | A dairy restricted diet |
| Age of Onset | Within days from birth |
| Prognosis | Early detection in the newborn period is the key to controlling symptoms. Long-term effects in untreated babies include severe mental retardation, cirrhosis of the liver, and death. About 75% of the untreated babies die within the first two weeks of life. |
| Disease | Sickle Cell Disease (SCD) |
| Metabolite | Thromboxane and Prostacyclin |
| Incidence | 1 in 500 African-American births, 1 in every 1,000 to 1 in 1,400 Hispanic-Americans |
| Symptoms | Painful events in the hands or feet, abdomen, back, and/or chest are the most common symptoms of sickle cell disease. This pain may last from hours to days. Most people with sickle cell disease experience anemia. Symptoms of anemia include feeling weak and tired. People with sickle cell disease can appear pale or washed out. Or they have a yellowish look to their skin and the whites of their eyes (jaundice). |
| Treatment | Pain medications (for sickle cell crises), drinking plenty of water daily or receiving fluid intravenously (to prevent and treat pain crises), blood transfusions (For anemia and to prevent stroke), and blood transfusions, are also used to dilute the HbS with normal hemoglobin to treat chronic pain, acute chest syndrome, splenic sequestration, and other emergencies. Other treatments include penicillin (to prevent infections) and folic acid (to help prevent severe anemia). Hydroxyurea is a medication that has recently been developed that may help reduce the frequency of pain crises and acute chest syndrome. It may also help decrease the need for frequent blood transfusions. However, the long-term effects of the medication are unknown. Also, a bone marrow transplant has proved to be effective in curing some persons with sickle cell disease. |
| Age of Onset | Less than 1 year |
| Prognosis | In the past, sickle-cell patients often died from organ failure between ages 20 and 40. Thanks to a better understanding and management of the disease, today patients can live into their 50s or beyond.Causes of death include organ failure and infection. Some people with the disease experience minor, brief, infrequent episodes. Others experience severe, long-term, frequent episodes with many complications |